BD Pompa a siringa Alaris™ PK Plus MK4 Istruzioni per l'uso
- Tipo
- Istruzioni per l'uso

Pompa a siringa Alaris™
PK Plus
MK4
Modello: 8005TIG03
Istruzioni per l’uso
it

BDDF00638 Edizione 2
1/56
Pompa a siringa Alaris™ PK Plus
Sommario
Pagina
Introduzione ..............................................................................................2
Informazioni sul manuale ..................................................................................3
Descrizione del modello TCI ...............................................................................4
Creazione di un set di dati .................................................................................7
Caratteristiche della pompa a siringa Alaris PK Plus .........................................................8
Comandi e indicatori ......................................................................................9
Definizione dei simboli ...................................................................................10
Funzioni principali del display ............................................................................11
Precauzioni di esercizio ..................................................................................13
Preparazione all’impiego .................................................................................16
Caricamento della siringa .................................................................................18
Avvio della pompa .......................................................................................21
Funzioni di base ..........................................................................................23
Operazioni da eseguire durante l’uso .....................................................................25
Allarmi e avvisi ...........................................................................................27
Messaggi ................................................................................................31
Opzioni di configurazione ................................................................................32
Specifiche tecniche .......................................................................................37
Siringhe riconosciute ....................................................................................40
Prodotti associati .........................................................................................41
Set di prolunga compatibili ..............................................................................42
Manutenzione ...........................................................................................45
Limiti della pressione di occlusione .......................................................................47
Specifiche IrDA, RS232 e Chiamata Infermiere .............................................................48
Curve a tromba e di avvio ................................................................................50
Profili in modalità TCI .....................................................................................51
Prodotti e parti di ricambio ...............................................................................54
Cronologia del documento ...............................................................................55
Contatti ..................................................................................................56

BDDF00638 Edizione 2
2/56
Pompa a siringa Alaris™ PK Plus
Introduzione
Introduzione
Queste Istruzioni per l'uso possono essere utilizzate con la Pompa a siringa Alaris™ PK Plus MK4
w
Le pompe possono essere identificate come versione MK4
grazie all'apposita etichetta posta sulla parte posteriore,
vedere l'immagine a destra, o controllando che la versione del
software sia 3.4.x o superiore durante l'accensione.
La pompa a siringa Alaris PK Plus (di seguito denominata "Pompa" nel testo) è un dispositivo di infusione utilizzabile per la
somministrazione di anestetici. Il software incorporato nella pompa comprende modelli predittivi farmacocinetici a tre compartimenti
eoffre quattro modalità operative:
1. Infusione continua (ml/h)
2. Anestesia totale endovenosa (TIVA)
– In questa modalità l’utente può scegliere la velocità di infusione e somministrare le dosi di bolo necessarie.
3. Anestesia totale per endovena (TIVA) con modalità predittiva TCI
– In questa modalità l’utente può scegliere la velocità di infusione e somministrare le dosi di bolo necessarie. Il modello
farmacocinetico viene utilizzato per stimare la concentrazione nel plasma e nel sito effettore.
4. Modo TCI
• Anestesia totale endovenosa controllata (TCI).
– In questa modalità l’utente seleziona la concentrazione target desiderata nel plasma e il modello farmacocinetico viene usato
per calcolare le velocità di infusione necessarie per raggiungere la concentrazione specificata. Un display grafico mostra il
trend della concentrazione stimata nel plasma e nel sito effettore nel tempo.
• Anestesia totale endovenosa controllata con target al sito effettore (TCI).
– In questa modalità l’utente imposta la concentrazione target nel sito effettore desiderata e il modello farmacodinamico viene
usato per calcolare le velocità di infusione necessarie per raggiungere la concentrazione specificata. Un display grafico mostra
il trend della concentrazione stimata nel plasma e nel sito effettore nel tempo.
La pompa a siringa Alaris PK Plus è caratterizzata da un'interfaccia di facile uso che visualizza la velocità di infusione, la dose totale di
farmaco somministrata e le concentrazioni stimate nel plasma e nel sito effettore, consentendo all'utente di conformarsi alle prescrizioni
sui farmaci applicabili nei singoli paesi.
Destinazione d'uso prevista
La pompa a siringa Alaris PK Plus è destinata all'uso da parte di personale medico per controllare la velocità di infusione e il volume.
Condizioni d'uso
La pompa a siringa Alaris PK Plus deve essere utilizzata solo da personale clinico competente nell'uso di pompe a siringa automatiche
nella gestione dei cateteri intravenosi.
L'uso della pompa a siringa Alaris PK Plus NON limita la responsabilità dell'anestesista incaricato della somministrazione dei farmaci.
È indispensabile che gli utenti della pompa a siringa Alaris PK Plus siano a conoscenza di tutta la letteratura disponibile per il modello
utilizzato con un farmaco e che rispettino i limiti di velocità e dosaggio prescritti. Pur essendo note, le interazioni farmacocinetiche e
farmodinamiche tra i farmaci anestetici non vengono utilizzate per calcolare la concentrazione nel plasma e nel sito effetto.
L'utente deve essere stato debitamente addestrato e rispettare tutte le raccomandazioni riportate in questo documento.
In particolare, l'utente deve essere a conoscenza del fatto che l'avvio della pompa in modalità TCI provoca l'infusione automatica di
una dose di bolo pre-calcolata, seguita da un'infusione, fino al raggiungimento della concentrazione target selezionata. I calcoli dei
parametri iniziali vengono visualizzati sul display prima dell'avvio dell'infusione. È pertanto essenziale che l'utente tenga conto delle
caratteristiche del paziente e che la velocità di infusione o la concentrazione nel plasma selezionate siano conformi alle informazioni
sulle prescrizioni dei farmaci in uso nei singoli paesi.
BD ha verificato la precisione dell'implementazione del modello matematico nonché la precisione di somministrazione della pompa.
Perinformazioni sui dati tecnici e sulla precisione di somministrazione della pompa, consultare la sezione 'Profili in modalità TCI'.
I singoli farmaci sono associati a modelli dedicati. Ciascun modello comprende un set di parametri farmacocinetici standard che
possono essere selezionati e usati dal modello a 3 comparti incorporato nella Pompa a siringa Alaris PK Plus (a condizione che l'uso del
farmaco sia consentito in modalità TCI).
Il Diprivan di ASTRA-ZENECA è l'unica formulazione a base di propofol che può essere utilizzata in modalità TCI in conformità ai dati sulle
prescrizioni dei farmaci. La pompa comprende anche il modello "Marsh", utilizzabile per il calcolo delle velocità di infusione del Diprivan
e delle concentrazioni nel plasma e nel sito effetto.
Se si utilizzano il Remifentanil ed il Sufentanil in modalità TCI, le velocità di infusione vengono calcolate utilizzando i modelli "Minto"
e"Gepts".

BDDF00638 Edizione 2
3/56
Pompa a siringa Alaris™ PK Plus
Informazioni sul manuale
w
BD non garantisce l'accuratezza costante del sistema se utilizzato con siringhe di altri produttori, come riportato
nella tabella 'Siringhe riconosciute'. I produttori possono cambiare senza preavviso le specifiche delle siringhe
influenzando in modo significativo la precisione del sistema.
Indicazioni
La pompa a siringa Alaris PK Plus è indicata per la somministrazione di anestetici
Controindicazioni
La pompa a siringa Alaris PK Plus è controindicata per:
• terapie enterali
• terapie di infusione epidurali
Informazioni sul manuale
Acquisire familiarità con tutte le caratteristiche della pompa a siringa Alaris PK Plus descritte in questo manuale prima di usarla.
Tutte le illustrazioni contenute in questo manuale si riferiscono a impostazioni e valori tipici, utilizzabili per programmare le funzioni della
pompa. Questi valori e queste impostazioni vengono forniti solo a titolo di esempio. La velocità di infusione minima, se indicata, siriferisce
a una velocità di infusione nominale di 1,0 ml/h, mentre quella intermedia si riferisce a una velocità di infusione nominale di 5,0 ml/h.
Perinformazioni complete sul range delle velocità di infusione, le impostazioni e i valori, vedere la sezione 'Specifiche tecniche'.
w
Conservare il presente Manuale per consultazioni future nel corso della vita utile della Pompa.
È importante fare riferimento esclusivamente alla versione più recente delle istruzioni per l'uso e del manuale tecnico
di servizio per i prodotti BD utilizzati. Tali documenti sono consultabili all'indirizzo bd.com. Le copie cartacee delle
istruzioni per l'uso possono essere ottenute gratuitamente contattando il rappresentante BD di zona. Una volta
effettuato l'ordine, verrà indicato un tempo di consegna approssimativo.
Convenzioni utilizzate in questo manuale
GRASSETTO Usato per i nomi delle schermate, i comandi software, i controlli e gli indicatori contenuti nel manuale,
adesempio, Indicatore di batteria, SPURGO, pulsante ON/OFF.
'Virgolette singole' Utilizzate per indicare i riferimenti incrociati ad altre sezioni del manuale.
Corsivo Usato quando si fa riferimento ad altri documenti o manuali nonché per dare enfasi.
Simbolo di avvertenza. Un'avvertenza è un'affermazione che segnala all'utente la possibilità di lesioni
personali anche mortali o altre gravi reazioni avverse associate all'uso o all'uso improprio di una pompa.
w
Simbolo di attenzione. Una nota di attenzione è un'affermazione che segnala all'utente la possibilità di
un problema con una pompa, associato al suo uso o all'uso improprio. Tali problemi possono includere
malfunzionamento della pompa, guasto alla pompa, danno alla pompa o ad altre proprietà. Un'attenzione
include le precauzioni da adottare per evitare il pericolo.

BDDF00638 Edizione 2
4/56
Pompa a siringa Alaris™ PK Plus
Descrizione del modello TCI
Descrizione del modello TCI
Il rapporto dose/risposta può essere diviso in tre parti: il rapporto tra la dose somministrata e la concentrazione nel plasma (fase
farmacocinetica), il rapporto tra la concentrazione nell’organo in cui il farmaco ha effetto e l’effetto clinico (fase farmacodinamica), e la
combinazione tra farmacocinetica e farmacodinamica. Lo scopo principale di somministrare un farmaco ad una dose specifica risiede
nella necessità di ottenere un effetto clinico specifico, che si verifica solo somministrando una dose terapeutica specifica nel sito di
azione (recettore).
DISTRIBUZIONE DOSE ESCREZIONE METABOLISMO
CONCENTRAZIONE NEL PLASMA
CONCENTRAZIONE NELLA BIOFASE
INTERAZIONE FARMACO-RECETTORE
EFFETTO
Fig. 1: rappresentazione schematica della farmacocinetica e dei processi dinamici che determinano il rapporto tra la dose somministrata
el'intensità dell'effetto risultante di un farmaco. Il rapporto tra la dose e la concentrazione del farmaco nel plasma e nella biofase (sito effettore)
è influenzato da alcuni fattori farmacocinetici come la distribuzione, il metabolismo e/o l’escrezione. Il farmaco interagisce con il recettore nella
biofase, provocando l'effetto farmacologico.
1
Fino a non molto tempo fa, gli anestetici somministrati per endovena, utilizzati per l’induzione o il mantenimento dello stato di
anestesia, venivano somministrati manualmente o per mezzo di pompe di infusione. In quest’ultimo caso la quantità di anestetico da
somministrare con la pompa veniva calcolata dall’anestesista in base al peso del paziente. Tuttavia, con questi metodi non è possibile
effettuare alcuna misurazione della concentrazione in linea. Inoltre, per calcolare le equazioni con più esponenti, richieste per stimare
leconcentrazioni, sono necessari sistemi informatici molto potenti. La sperimentazione di Kruger-Thiemer
2
e Schwilden et al.
3
ha
portato, negli anni '80 e '90, allo sviluppo del modello TCI, in quanto i progressi nella tecnologia informatica hanno reso possibile la
stima delle concentrazioni dei farmaci durante il processo.
Il comportamento farmacocinetico di molti anestetici può essere descritto matematicamente utilizzando un modello a 3 compartimenti:
un compartimento centrale (V1), un compartimento ricco di vasi sanguigni (V2) e un compartimento povero di vasi sanguigni (V3).
Il trasferimento del farmaco tra i vari compartimenti (distribuzione) viene descritto dalle costanti di velocità (k
12
, k
21
, k
31
e k
13
) o di
clearance. Il metabolismo del farmaco viene descritto dalla costante della velocità k
10
(Fig. 2). Lo scopo del modello TCI consiste
nell’utilizzare i modelli farmacocinetici per calcolare le velocità di infusione richieste per ottenere la concentrazione desiderata del
farmaco nel plasma. In questo caso, tuttavia, non si specifica una velocità di infusione, ma una concentrazione "target" sulla base di
considerazioni cliniche. Nel caso specifico della concentrazione target nel compartimento plasma si parla di "TCI a loop aperto riferita al
plasma". Nel caso specifico di concentrazione target riferita al compartimento effetto, si parla di "TCI a loop aperto riferito al sito effetto".
Compartimento
Compartimento Compartimento
Clearance cl1
cl2 cl3
Fig. 2: rappresentazione schematica del modello a tre compartimenti usato per le infusioni basate sulle concentrazioni target.
Per gli anestetici, il sito effettore (o biofase) non è il plasma
4
ma il cervello, dove non è possibile misurare direttamente le concentrazioni.
Fino ai primi anni ‘90 si riteneva che il bilanciamento della concentrazione nel sangue/cervello fosse virtualmente immediato. Per questo
tutti i modelli TCI iniziali utilizzavano come target il plasma. Per molti farmaci il rapporto tra la concentrazione nel plasma e l’effetto
clinico veniva generalmente descritto in termini di Cp50 o Cp95 (che rappresentavano le concentrazioni richieste per ottenere un effetto
clinico specifico rispettivamente nel 50% e nel 95% dei pazienti). Per un esempio, consultare Ausems et al.
5
Nel corso degli anni 90 divenne, tuttavia, sempre più evidente che variazioni della concentrazione nel plasma provocavano un
ritardo temporale nel bilanciamento tra le concentrazioni nel plasma e nel sito effettore. Poiché l'effetto clinico varia parallelamente
alla concentrazione nel sito effettore, per molti farmaci la velocità di distribuzione verso/dal sito di azione può essere caratterizzata
dall'effetto del farmaco nel tempo
6,7
. Ciò significa che l’effetto può essere trasferito alle concentrazioni, con la conseguente possibilità
di adottare un approccio quantitativo. La concentrazione nel sito di azione è chiamata "concentrazione nel sito effettore" e il
compartimento corrispondente
8
(vedere Fig. 3) viene detto "compartimento del sito effettore". Poiché la quantità effettiva di farmaco
che arriva al cervello è molto limitata, il compartimento del sito effettore ha un volume praticamente nullo e una costante di velocità
k
1e
trascurabile. La costante di velocità k
eo
può essere invece usata per descrivere la velocità di bilanciamento tra i compartimenti del
plasma e del sito effettore.

BDDF00638 Edizione 2
5/56
Pompa a siringa Alaris™ PK Plus
Descrizione del modello TCI
La possibilità di individuare la costante k
eo
di molti agenti ha permesso di conoscere la concentrazione target nel sito effettore. Il
modello TCI consente prima di calcolare il profilo della concentrazione nel plasma richiesto per ottenere la concentrazione target nel
sito effettore il più rapidamente possibile, quindi le velocità di infusione richieste per ottenere un profilo di concentrazione nel plasma
specifico (Fig 3). Il rapporto tra la concentrazione nel sito effettore e nel plasma genera una dose di induzione maggiore seguita da una
pausa nell’infusione che consente al plasma di equilibrarsi in base alla concentrazione nel sito effettore.
V2
Compartimento
periferico
V1
Compartimento
centrale
V3
Compartimento
periferico
Somministrazione
k
21
k
12
k
13
k
31
k
10
k
eo
Compartimento
effetto
Clearance cl1
cl2 cl3
Fig. 3: Rappresentazione schematica del rapporto concentrazione/effetto.
Le pompe di infusione TCI consentono di controllare in modo ottimale l’anestesia, soprattutto se i tre elementi descritti in precedenza
sono stati calcolati e descritti in modo preciso. In primo luogo è importante che il modello utilizzato per la pompa funzioni in modo
preciso. I modelli usati per la pompa a siringa Alaris PK Plus sono stati ampiamente convalidati e certificati. In secondo luogo, il set di
parametri farmacocinetici di un farmaco specifico utilizzato dal modello computerizzato deve corrispondere al profilo farmacocinetico
del paziente (tenere presente che i modelli descritti nella letteratura sono basati su dati di "popolazione" e sono validi per un paziente
"medio", ma non tengono conto della variabilità farmacocinetica tra i pazienti). Infine, è necessario conoscere bene la farmacodinamica
della molecola somministrata per permettere all’utente di selezionare la concentrazione nel plasma e nel sito effetto, necessaria per
ottenere l’effetto desiderato. Per la maggior parte degli anestetici, la farmacodinamica varia considerevolmente da paziente a paziente,
quindi è indispensabile allineare i dati farmacodinamici riferiti al campione di popolazione ai dati clinici del paziente per stabilire la
sensibilità specifica del paziente al farmaco e consentire l’effetto titolazione se richiesto.
Nota: i parametri specifici al modello sono disponibili nella sezione 'Descrizione del modello TCI' o direttamente sulla Pompa,
consultabili premendo il tasto Info durante la selezione del farmaco. Prima di utilizzare la modalità TCI, è indispensabile anche
controllare le informazioni fornite con il farmaco per verificare che l’uso di questa modalità sia permesso nel proprio paese di
residenza.
Bibliografia:
1. Danhof M: Does variability explain (all) variability in drug effects ?, Topics in pharmaceutical science. Edited by Breimer DD, Crommelin DJA, Midha KK. Noordwijk, Amsterdam
Med. Press BV, 1989, pp 573-586
2. Kruger-Theimer E: Continuous intravenous infusion and multicompartment accumulation. Eur J Pharmacol 1968; 4: 317-324
3. Schwilden H: A general method for calculating the dosage scheme in linear pharmacokinetics. Eur J Clin Pharmacol 1981; 20: 379-86
4. Shafer SL: Towards optimal intravenous dosing strategies. Seminars in Anesthesia 1993; 12: 222-234
5. Ausems ME, Hug CC, Jr., Stanski DR, Burm AG: Plasma concentrations of alfentanil required to supplement nitrous oxide anesthesia for general surgery. Anesthesiology 1986;
65: 362-73
6. Schnider TW, Minto CF, Stanski DR: The effect compartment concept in pharmacodynamic modelling. Anaesthetic Pharmacology Review 1994; 2: 204-213
7. Shafer SL: Principles of pharmacokinetics and pharmacodynamics., Principles and practice of anesthesiology. 2nd Edition. Edited by Longnecker DE, Tinker JH, Morgan GE.
New York, Mosby-Year Book, 1998, pp 1159- 1210
8. Shafer SL, Gregg KM: Algorithms to rapidly achieve and maintain stable drug concentrations at the site of drug effect with a computer-controlled infusion pump. J
Pharmacokinet Biopharm 1992; 20: 147-69
Precauzioni relative all’uso del modello TCI
Al primo avvio i modelli farmacocinetici/farmacodinamici delle pompe a siringa Alaris PK Plus sono reimpostati a zero. Pertanto, se la
pompa viene spenta mentre l’infusione è in corso, tutti i dati farmacocinetici/farmacodinamici vengono cancellati. Lo spegnimento e il
riavvio della pompa ed il proseguimento dell’infusione in queste condizioni possono provocare una sovrainfusione, poiché al paziente è
già stata somministrata una quantità significativa di farmaco. Quindi, è sempre preferibile evitare di riavviare la pompa in modalità TCI.

BDDF00638 Edizione 2
6/56
Pompa a siringa Alaris™ PK Plus
Descrizione del modello TCI
Modelli farmacocinetici e relativi parametri nelle pompe a siringa Alaris PK Plus
Farmaco: Diprivan Modello: Marsh (con correzione del peso)
Limite di età: minimo 16 anni
Unità di misurazione della concentrazione nel plasma: µg/ml
Concentrazione max nel plasma: 15 µg/ml
Vc = 0,228 x massa (litri x kg
-1
)
k
10
= 0,119 minuti
-1
k
12
= 0,112 minuti
-1
k
13
= 0,0419 minuti
-1
k
21
= 0,055 minuti
-1
k
31
= 0,0033 minuti
-1
k
eo
= 0,26 minuti
-1
Riferimenti in letteratura: Marsh et al.: Brit J Anaesth 1991, 67, 41-48
Farmaco: Remifentanil Modello: Minto
Limite di età: minimo 12 anni
Unità di misurazione della concentrazione nel plasma: ng/ml
Concentrazione max: 20 ng/ml
Vc = 5,1 - 0,0201 x (età-40) + 0.072 x (lbm-55)
V2 = 9,82 - 0,0811 x (età - 40) + 0,108 x (lbm - 55)
V3 = 5,42
cl1 = 2,6 - 0,0162 x (età - 40) + 0,0191 x (lbm - 55)
cl2 = 2,05 - 0,0301 x (età - 40)
cl3 = 0,076 -0,00113 x (età - 40)
k
10
= cl1 / Vc
k
12
= cl2 / Vc
k
13
= cl3 / Vc
k
21
= cl2 / V2
k
31
= cl3 / V3
k
eo
= 0,595 - 0,007 x (età - 40)
Riferimenti in letteratura: Minto et al.: Anesthesiology 1997, 86, 10 - 33
Farmaco: Sufentanil Modello: Gepts (senza correzione del peso)
Limite di età: minimo 12 anni
Unità di misurazione della concentrazione nel plasma: ng/ml
Concentrazione max: 2 ng/ml
Vc = 14,3 l
k
10
= 0,0645 minuti
-1
k
12
= 0,1086 minuti
-1
k
13
= 0,0229 minuti
-1
k
21
= 0,0245 minuti
-1
k
31
= 0,0013 minuti
-1
Riferimenti in letteratura: Gepts et al.: Anesthesiology 1995, 83, 1194 -1204
Elementi aggiuntivi:
valore k
eo
calcolato con effetto di tempo al picco pari a 5,6 minuti (k
eo
= 0,17559 minuti
-1
) (riferimento: Shafer et al Anesthesiology.
1991Jan;74(1):53-63)

BDDF00638 Edizione 2
7/56
Pompa a siringa Alaris™ PK Plus
Creazione di un set di dati
Creazione di un set di dati
Per utilizzare a pieno la pompa a siringa Alaris PK Plus è necessario sviluppare, revisionare, approvare, rilasciare, caricare e verificare un
Data Set attenendosi alla procedura descritta di seguito. Consultare le istruzioni d’uso del software Alaris PK Editor (1000CH00016) per
maggiori dettagli e precauzioni operative.
1. Creare elenchi master (usando il software Alaris PK Editor)
• Farmaci master* Elenco di nomi di farmaci e concentrazioni standard. Possono essere destinati
all’uso in modalità TIVA o possono avere un modello PK/PD associato per l’uso
in modalità TCI.
• Libreria siringhe Alaris PK Configura le siringhe abilitate all’uso.
2. Creare il profilo (usando il software Alaris PK Editor)
• Farmaci del profilo* Farmaci e concentrazioni per questo profilo con valori predefiniti, limiti minimi
e massimi e target, nonché livelli di occlusione.
• Configurazione della pompa** Impostazioni e opzioni generali di configurazione della pompa.
3. Revisionare, approvare e pubblicare (usando il software Alaris PK Editor)
• Revisione e approvazione Resoconto completo sul Data Set da stampare, revisionare e firmare a riprova
dell’approvazione da parte di una persona autorizzata in base al protocollo
ospedaliero. Il documento firmato deve essere conservato al sicuro per
un’eventuale consultazione durante la procedura di verifica.
• Pubblicazione Lo stato del Data Set deve essere impostato su Rilasciato (è richiesta una
password).
4. Caricare il Data Set nella pompa a siringa Alaris PK Plus (usando lo strumento di trasferimento di Alaris PK Editor)
5. Verificare il caricamento del Data Set
• Prima o unica verifica della pompa Una volta completato il caricamento, registrare il numero CRC (Cyclic
Redundancy Check) riportato sulla pompa a siringa Alaris PK Plus.
Scaricare il Data Set dalla pompa mediante il software Alaris PK Verification Tool.
Confrontare il Data Set scaricato con la stampa del Data Set approvata e
firmata. Il revisore deve firmare la stampa e annotare su di essa il numero CRC
ascopo di registrazione.
• Verifica successiva della pompa Successivamente, ogniqualvolta si caricano i set di dati, confrontare il numero
CRC sulla pompa con il numero CRC registrato sulla prima verifica della pompa.
6. Accendere la Pompa e verificare che nella schermata iniziale all'avvio siano visualizzati il nome e la versione corretti del set di dati.
Ora la pompa è pronta per l'uso.
w
* I parametri dei farmaci devono rispettare i protocolli vigenti e le informazioni prescritte.
Il trasferimento del Data Set deve essere effettuato esclusivamente da parte di personale qualificato.
** Vedere la nota importante nella sezione 'Opzioni di configurazione'.

BDDF00638 Edizione 2
8/56
Pompa a siringa Alaris™ PK Plus
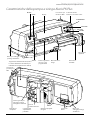
Caratteristiche della pompa a siringa Alaris PK Plus
Caratteristiche della pompa a siringa Alaris PK Plus
Indicatore allarme
luminoso con codice colore
Leva rilascio per
interfaccia MDI
Tasti di regolazione
a freccia e tasti
programmabili
Pinze blocca
stantuffo
Display
Morsetto blocca
siringa
ON/OFF
AVVIO
ATTESA
SPURGO/BOLO
SILENZIARE
PRESSIONI
OPZIONI
Gancio per cablaggio
prolunga di infusione
Impugnature
sagomate
Gancio per
cablaggio prolunga
di infusione
Leva rilascio
per interfaccia
MDI
Maniglia per
il trasporto
Porta di
comunicazione IR
Connettore
RS232
Morsetto ripiegabile
integrato
Connettore PE
equalizzazione
potenziali
Targa dei dati di funzionamento (vedere
la sezione 'Definizione dei simboli' per
informazioni sul significato dei simboli)
Camma girevole per
il fissaggio su barre
rettangolari orizzontali
I
n
t
e
r
f
a
c
c
i
a
M
D
I

BDDF00638 Edizione 2
9/56
Pompa a siringa Alaris™ PK Plus
Comandi e indicatori
Comandi e indicatori
Comandi:
Simbolo Descrizione
a
Pulsante ON/OFF - Premere il pulsante una sola volta per accendere la pompa. Tenere premuto il
pulsante per 3 secondi per spegnere la pompa.
Nota: è possibile spegnere la pompa (off) solo in determinate fasi di funzionamento, per informazioni
dettagliate vedere la sezione 'Procedura di spegnimento' in 'Opzioni di configurazione'.
Nota: i registri sono conservati in caso di interruzione del funzionamento, compreso in caso di
spegnimento della pompa o di interruzione imprevista dell'alimentazione.
b
Pulsante AVVIO - Premere questo pulsante per iniziare l’infusione. Il LED verde lampeggia mentre
l’infusione è in corso.
h
Pulsante ATTESA - Premere questo pulsante per sospendere l’infusione. Il LED giallo si accende quando
la pompa è in attesa.
R
Pulsante SILENZIARE - Premere questo pulsante per tacitare l’allarme per 2 minuti (l’intervallo è
configurabile). Per riattivare l'audio dell'allarme, premere il pulsante SILENZIARE una seconda volta.
Nota: solo allarme di attenzione.
– I due minuti di silenzio possono essere configurati mediante il software Alaris PK Editor.
– Quando l'allarme non è attivo, tenere premuto il pulsante fino a quando la pompa non
emette quattro segnali acustici a intervalli di 60 minuti..
i
Pulsante SPURGO/BOLO - Premere questo pulsante per utilizzare i tasti programmabili SPURGO o
BOLO. Per avviare la pompa, premere e tenere premuto il tasto programmabile.
Spurgare il set di prolunga durante la preparazione.
• La pompa è in attesa
• Il set di prolunga non è collegato al paziente
• Il volume infuso (VI) non viene incrementato
BOLO - Fluido o farmaco somministrato a un regime accelerato.
• La pompa sta eseguendo l’infusione
• Il set di prolunga è collegato al paziente
• Il VI viene incrementato
d
Pulsante OPZIONI - Premere questo pulsante per visualizzare le funzioni opzionali (vedere 'Funzioni di
base').
e
Pulsante PRESSIONI - Usare questo pulsante per visualizzare i trend della pressione di pompaggio
eillivello degli allarmi.
f
FRECCE - I tasti a freccia doppia o singola consentono di aumentare o diminuire, più o meno
rapidamente, i valori visualizzati sul display.
g
TASTI PROGRAMMABILI VUOTI - Utilizzare questi tasti con i messaggi di richiesta visualizzati sul
display.
Indicatori:
Simbolo Descrizione
j
Indicatore BATTERIA - Se acceso, indica che la pompa è alimentata dalla batteria interna. Se lampeggia,
indica che la batteria ha un’autonomia di carica inferiore a 30 minuti.
S
Indicatore ALIMENTAZIONE IN C. A. - Se acceso, indica che la pompa è collegata alla rete CA e che la
batteria è in carica.

BDDF00638 Edizione 2
10/56
Pompa a siringa Alaris™ PK Plus
Definizione dei simboli
Definizione dei simboli
Simboli delle etichette
Simbolo Descrizione
Consultare la documentazione allegata.
x
Connettore PE (per equalizzazione potenziale)
y
Connettore RS232/Chiamata Infermiere (opzionale)
l
Dispositivo di tipo CF a prova di defibrillatore (grado di protezione dalle scariche elettriche)
IP32
Protezione contro spruzzi diretti di acqua fino a 15° dalla verticale e protezione contro oggetti solidi più grandi
di2,5 mm.
Nota: la classificazione IP33 si applica se è installato il kit di protezione per cavo di alimentazione CA, numero
di parte 1000SP01294.
r
Corrente alternata
Dispositivo conforme ai requisiti della Direttiva CE 93/42/CEE, secondo la rettifica 2007/47/CE.
T
Data di fabbricazione
t
Produttore
U
Non indicato per lo smaltimento come rifiuto urbano
W
Caratteristiche elettriche dei fusibili
+40°C
0°C
Intervallo temperatura di esercizio - la pompa può essere utilizzata in un intervallo compreso tra 0 °C e 40 °C.

BDDF00638 Edizione 2
11/56
Pompa a siringa Alaris™ PK Plus
Funzioni principali del display
Funzioni principali del display
Modo TIVA
Stato della
pompa
Nome del farmaco
econcentrazione
Dati pressione
Velocità del flusso
e di dosaggio
Dosaggio e
volume infuso
Funzioni
opzionali
Modo TCI
CONFIRM TIME
Stato della pompa
Nome del farmaco
econcentrazione
Concentrazione
plasmatica
Target
concentrazione
plasmatica
Velocità
induzione
iniziale
Velocità di
mantenimento
iniziale
Pausa prima della
manutenzione
Durata
induzione
Tempo di
induzione
Dosaggio
induzione
iniziale
Volume
induzione
iniziale
Modo TCI - Schermata ALTRO
Premere il tasto programmabile ALTRO per visualizzare ulteriori informazioni:
BMI 21.6
Parametri paziente Tempo per completamento
infusione a velocità corrente
Volume e dose
infusi
Tempo
trascorso
Nome farmaco
emodello
Tempo di
decremento
Concentrazione
decremento
Premere il tasto programmabile INDIETRO per tornare alla schermata TCI. Il display torna a visualizzare automaticamente la schermata
TCI dopo circa 20 secondi.

BDDF00638 Edizione 2
12/56
Pompa a siringa Alaris™ PK Plus
Funzioni principali del display
Icone sul display
Simbolo Descrizione
l
TEMPO RIMANENTE - Indica quanto tempo intercorre prima della sostituzione della siringa.
N
Icona BATTERIA - Indica il livello di carica della batteria e consente di stabilire se la batteria deve essere
ricaricata oppure ricollegata all'alimentazione CA.
Nota: può essere abilitata/disabilitata per mezzo del software Alaris Editor
C
Dose per la fase di induzione (visualizzato sulla schermata di conferma del protocollo)
D
Durata della fase di induzione (visualizzata sulla schermata di conferma del protocollo)
E
Durata dell’infusione in bolo automatico (visualizzata sulla schermata di impostazione del bolo)
F
Dosaggio per la fase di mantenimento (visualizzato sulla schermata di conferma del protocollo)
Soft Alert - Indica che la pompa sta funzionando ad una velocità superiore (rivolta verso l’alto) o inferiore
(rivolta verso il basso) ad un Soft Alert. (Il numero delle frecce varia a seconda della lunghezza del nome del
farmaco).
Hard Limit - Indica che l’impostazione selezionata supera o è inferiore al valore di un Soft Alert oppure non
èconsentita perché supera un Hard Limit.
RIDUZIONE DELLA CONCENTRAZIONE - Stato dell’infusione che indica che la concentrazione target è
inferiore alla concentrazione corrente.

BDDF00638 Edizione 2
13/56
Pompa a siringa Alaris™ PK Plus
Precauzioni di esercizio
Precauzioni di esercizio
Siringhe monouso e set di prolunga
m
• La pompa a siringa Alaris PK Plus è stata calibrata per l’uso con siringhe monouso. Per garantire il
funzionamento ottimale e corretto, usare solo siringhe Luer lock in 3 pezzi del tipo specificato sulla
pompa o consigliato nel manuale. L’uso di siringhe o set di prolunga diversi da quelli consigliati può
deteriorare il funzionamento della pompa e la precisione dell’infusione.
n
• L’installazione impropria della siringa sulla pompa o la sua rimozione dalla pompa prima dell’isolamento
del tubo di prolunga dal paziente possono provocare problemi come flusso incontrollato o sifonaggio.
Per isolare il tubo di prolunga può essere necessario chiudere un rubinetto sul tubo collegato al paziente
o usare un morsetto o clamp arrestaflusso.
o
• Fissare il set di prolunga alla Pompa utilizzando l'apposito gancio sul retro della Pompa. Questo
accorgimento garantisce un'ulteriore protezione in caso di distacco accidentale della siringa dalla Pompa.
• L’uso combinato di più apparecchiature e/o strumenti con set di prolunga e altri tipi di tubi, come i
rubinetti a 3 vie, può influire sulle prestazioni della pompa e comportare la necessità di monitorare con
attenzione la pompa.
• Si deve sempre bloccare con un morsetto o isolare in altro modo la tubazione di mandata al paziente
prima di rilasciare il morsetto o rimuovere la siringa dalla pompa. Qualsiasi mancanza in questo senso
può causare una somministrazione involontaria.
Montaggio della pompa
• Quando su un paziente viene utilizzata più di una pompa, quelle contenenti farmaci critici e ad alto
rischio devono essere posizionate il più vicino possibile al livello del cuore del paziente onde evitare il
rischio di variazioni di flusso o sifonaggio.
• Il sollevamento della Pompa durante l'infusione può risultare in un bolo di soluzione infusa mentre
l'abbassamento della Pompa durante l'infusione può risultare in un ritardo nell'infusione (infusione
insufficiente).
I
• Non montare la pompa in posizione verticale con la siringa rivolta verso l’alto, per evitare di provocare
l’infusione dell’aria eventualmente contenuta nella siringa. Per evitare l’ingresso dell’aria, è indispensabile
controllare regolarmente l’avanzamento dell’infusione, le connessioni della siringa, il tubo di prolunga e le
connessioni del paziente, seguendo attentamente la procedura di adescamento descritta in questo manuale.
Ambiente operativo
• Gli ambienti di destinazione includono i reparti di terapia intensiva e le sale operatorie. È possibile
utilizzare la pompa in ambulanza. Accertarsi che la pompa sia correttamente collegata usando il morsetto
per asta in dotazione. La pompa è progettata per resistere a possibili sobbalzi e vibrazioni durante
l'utilizzo in ambulanza, in conformità con lo standard EN 1789. Se la pompa cade o subisce gravi danni,
predisporre un'attenta ispezione da parte di personale tecnico qualificato non appena possibile.
È possibile utilizzare la pompa se la temperatura rientra nell'intervallo specificato in base a quanto
riportato nel capitolo 'Specifiche tecniche' e sull'etichetta della pompa.
• Quando si utilizzano pompe per infusione con altri tipi di pompe o dispositivi che richiedono un accesso
vascolare, è necessario prestare la massima attenzione. Una variazione significativa di pressione all’interno
del sistema vascolare locale, indotta da pompe di questo tipo, può essere responsabile di un’errata
somministrazione di farmaci o liquidi. Questo tipo di pompe viene generalmente usato durante dialisi,
interventi di bypass o applicazioni cardiache assistite.
• La Pompa è progettata per essere impiegata in ambienti medici e ospedalieri, diversi da quelli residenziali,
che hanno accesso all'alimentazione CA monofase.
• La pompa non è progettata per essere usata in ambienti in cui sono presenti sostanze anestetiche
infiammabili contenenti miscele di aria o ossigeno o protossido d’azoto.
Pressione di esercizio
• Questa pompa è un dispositivo a pressione positiva, progettato per garantire una somministrazione di
liquidi molto precisa, con compensazione automatica della resistenza incontrata nel sistema di infusione.
• Il sistema di allarme della pressione di pompaggio non è concepito per rilevare o per offrire protezione
contro complicazioni endovenose che possano accadere.

BDDF00638 Edizione 2
14/56
Pompa a siringa Alaris™ PK Plus
Precauzioni di esercizio
Condizioni di allarme
J
• Molte condizioni di allarme rilevate da questa pompa interrompono l’infusione e generano allarmi visivi
e sonori. Pertanto è necessario effettuare controlli regolari per accertarsi che l’infusione stia procedendo
correttamente e non vi siano segnalazioni di allarme.
• In caso di interruzione dell'alimentazione il volume di allarme impostato verrà mantenuto, tuttavia
alcuni errori di sistema provocheranno la perdita delle impostazioni di allarme. Le nuove impostazioni
di allarme verranno memorizzate durante lo spegnimento dalla modalità di assistenza dopo la modifica.
Le impostazioni verranno perse se viene effettuato un avvio definito "cold-start", ma dovrebbero essere
preservate per i guasti che non richiedono il cold-start.
Pericoli
• L’uso della pompa in presenza di anestetici infiammabili può provocare esplosioni. In questo caso, occorre
adottare la massima cautela e posizionarla lontano da queste fonti.
A
• Tensione pericolosa: l'apertura o la rimozione dell'alloggiamento della Pompa può esporre l'utente
al rischio di scariche elettriche. Far eseguire tutte le operazioni di riparazione da personale tecnico
qualificato.
• Se la pompa viene collegata a una fonte di alimentazione esterna, è necessario usare sempre una linea di
distribuzione a tre conduttori (fase, neutro, terra). Se si hanno dubbi sull’integrità della protezione esterna
del conduttore, o sulla sua installazione, azionare la pompa a batteria.
V
• Non aprire la copertura di protezione dell’interfaccia RS232/Chiamata Infermiere quando non è in uso.
Adottare tutte le misure necessarie per prevenire le scariche elettrostatiche al momento del collegamento
dell’interfaccia RS232/Chiamata Infermiere. Il contatto con i pin dei connettori può rendere nulla la
protezione contro le scariche elettrostatiche. È consigliabile far eseguire tutte le operazioni da personale
debitamente qualificato.
L
• In caso di caduta accidentale della pompa, presenza di condensa eccessiva, perdite di liquidi, umidità
o temperatura elevata o se si sospetta che possa aver subito danni, interromperne immediatamente
l'uso e farla ispezionare da personale di assistenza qualificato. Per trasportare o conservare la pompa,
usare sempre l’imballaggio originale, se possibile, e rispettare i limiti di temperatura, umidità e pressione
indicati nella sezione 'Specifiche tecniche' e sull'imballaggio esterno.
• Il software fornito con la pompa include i limiti e i parametri di configurazione. L’adeguatezza dei
limiti, lacompatibilità dei farmaci e le prestazioni di ogni pompa devono essere verificate da personale
qualificato come parte dell’intera procedura di infusione. I potenziali pericoli includono interazioni tra
farmaci, velocità di somministrazione e allarmi di pressione inadeguati.
• Le pompe a siringa Alaris possono essere modificate o alterate solo previa autorizzazione odisposizione
di BD. Qualsiasi utilizzo delle Pompe a siringa Alaris eventualmente alterate o modificate senza
previa istruzione da parte di BD è a esclusivo rischio dell'utente e BD non fornisce alcuna garanzia o
approvazione per qualsiasi Pompa a siringa Alaris modificata o alterata in tal modo. La garanzia del
prodotto BD non si applica in caso di danni, usura precoce, malfunzionamenti o altre anomalie causati
damodifiche o alterazioni non autorizzate della Pompa a siringa Alaris.
• Tutte le pompe in una singola di area cura devono essere configurate con gli stessi toni di allarme per
evitare confusione per l'utente.

BDDF00638 Edizione 2
15/56
Pompa a siringa Alaris™ PK Plus
Precauzioni di esercizio
Compatibilità e interferenze elettromagnetiche
M
• Questa pompa è protetta contro gli effetti derivanti dalle interferenze esterne, comprese le emissioni
in radiofrequenza ad alta energia, i campi magnetici e le scariche elettrostatiche (ad esempio quelle
generate da apparecchiature elettrochirurgiche e di cauterizzazione, motori di grandi dimensioni,
radio portatili, telefoni cellulari e così via) ed è progettata per garantire la sicurezza in presenza di livelli
eccessivi di interferenza.
• Apparecchiature di radioterapia: non utilizzare la Pompa in prossimità di eventuali apparecchiature di
radioterapia. I livelli di radiazioni generati dalle apparecchiature di radioterapia, come gli acceleratori
lineari, possono alterare gravemente il funzionamento della pompa. Consultare le raccomandazioni
del produttore per conoscere la distanza di sicurezza e le altre precauzioni necessarie. Per ulteriori
informazioni, contattare il rappresentante BD di zona.
MR
• Apparecchiature per la risonanza magnetica (MRI): la Pompa contiene materiali ferromagnetici soggetti
a interferenze con il campo magnetico generato dalle apparecchiature MRI. La pompa non può quindi
essere considerata intrinsecamente compatibile con MRI. Se non è possibile evitare di utilizzare la Pompa
in ambiente MRI, BD consiglia vivamente di fissarla a distanza di sicurezza dal campo magnetico, fuori
dalla zona identificata come "Area ad accesso controllato", per evitare qualsiasi interferenza magnetica
dannosa per la Pompa o distorsione delle immagini MRI. La distanza di sicurezza dovrebbe essere stabilita
conformemente alle raccomandazioni del produttore in materia di interferenze elettromagnetiche
(EMI). Per ulteriori informazioni, fare riferimento al Manuale di supporto tecnico (TSM) del prodotto. In
alternativa, contattare il rappresentante BD di zona per ulteriori istruzioni.
• Accessori: non utilizzare alcun accessorio non raccomandato con la Pompa. La pompa è stata collaudata
ed è conforme alle relative dichiarazioni EMC solo con gli accessori consigliati. L'utilizzo di qualsiasi
accessorio, trasduttore o cavo diverso da quelli specificati da BD può determinare un aumento delle
emissioni o una riduzione dell'immunità della Pompa.
• Questa pompa è un dispositivo CISPR 11, Gruppo 1, di Classe A e utilizza energia in radiofrequenza solo
per le funzioni interne nella configurazione standard. Pertanto, le emissioni RF sono molto basse e non
interferiscono generalmente con le apparecchiature elettroniche installate nelle vicinanze. Tuttavia, la
pompa emette un certo livello di radiazioni elettromagnetiche che rientrano nei livelli specificati nelle
normative IEC/EN60601-1-2 e IEC/EN60601-2-24. Se la pompa interferisce con altre apparecchiature,
è necessario adottare idonee misure per minimizzare gli effetti, ad esempio installandola in un’altra
ubicazione o posizione.
K
• In alcuni casi la pompa può essere esposta a scariche elettrostatiche pari o superiori a 15kv o a radiazioni
in radiofrequenza pari o superiori a 10v/m. Se esposta a queste interferenze esterne, la pompa imposta
la modalità di sicurezza, arresta prontamente l’infusione e avverte l’utente con una serie di allarmi visivi e
acustici. Se la condizione di allarme riscontrata persiste anche dopo l'intervento dell'utente, si consiglia di
sostituire la Pompa e isolarla, in modo che possa essere ispezionata da personale di assistenza qualificato
(per ulteriori informazioni, consultare il manuale tecnico di servizio).

BDDF00638 Edizione 2
16/56
Pompa a siringa Alaris™ PK Plus
Preparazione all’impiego
Preparazione all’impiego
Installazione iniziale
w
Prima di usare la pompa, leggere attentamente le istruzioni per l’uso riportate in questo manuale.
1. Verificare che la pompa sia integra, non danneggiata e che la tensione specificata sull’etichetta sia compatibile con quella della rete
di alimentazione CA utilizzata.
2. Articoli compresi:
• Pompa a siringa Alaris PK Plus
• CD per l’utente (istruzioni per l’uso)
• Istruzioni per l'uso in formato elettronico
• Cavo di alimentazione CA (come richiesto)
• Imballo di protezione
3. Lasciare collegata la Pompa all'alimentazione CA per almeno 2½ ore per caricare completamente la batteria interna (verificare che
ilsimbolo S si illumini).
Selezione della lingua
1. All’avvio iniziale, viene visualizzata la schermata Selezione lingua.
2. Selezionare la lingua dall’elenco visualizzato premendo i tasti
f.
3. Premere il tasto programmabile OK per confermare la selezione.
w
• La pompa viene alimentata automaticamente dalla batteria interna se non è collegata alla rete di alimentazione
CA al momento dell’accensione.
• In caso di malfunzionamento della pompa, riporla nell'imballaggio di protezione originale, se possibile,
econtattare il personale di assistenza qualificato per farla ispezionare.

BDDF00638 Edizione 2
17/56
Pompa a siringa Alaris™ PK Plus
Preparazione all’impiego
Non montare la pompa con l'ingresso di alimentazione CA o la siringa orientati verso l'alto, per evitare che l'eventuale
fuoriuscita di liquido comprometta la sicurezza elettrica o provochi l'infusione di aria eventualmente contenuta nella
siringa.
Installazione del morsetto
Il morsetto per palo, già montato sul retro della pompa, garantisce un
fissaggio sicuro dei dispositivi per somministrazione endovenosa ai pali
verticali con diametro compreso tra 15 e 40 mm.
*
*
Incavo
1. Tirare il morsetto ripiegabile verso l’esterno e allentarlo, lasciando
uno spazio sufficiente per l’inserimento del palo.
2. Montare la pompa sul palo e serrare il morsetto fino a fissarla
saldamente.
w
Verificare che il morsetto per palo sia ripiegato e inserito
nell’apposito incavo sul retro della pompa prima di
collegare la pompa a una Docking Station/Workstation*
o se si prevede di non usarla.
Non montare la pompa in una posizione che appesantisca
o renda instabile l’eventuale stativo per infusione.
w
Prima di ogni uso, verificare che il morsetto per palo:
• non mostri segni di usura eccessiva,
• non mostri segni di movimento eccessivamente allentato in posizione estesa di montaggio.
Se si osservano questi segni, interrompere l'uso della pompa e farla esaminare da personale di assistenza qualificato.
Installazione su Docking Station/Workstation* o su barra normalizzata
La camma girevole può essere montata sulla barra rettangolare della Docking Station/Workstation*.
1. Allineare la camma girevole sul retro della pompa con la barra rettangolare della Docking Station/Workstation* o con la barra
normalizzata.
2. Mantenere la pompa in posizione orizzontale, quindi premerla a fondo contro la barra rettangolare o la barra normalizzata.
3. La Pompa deve scattare in posizione quando viene fissata alla barra.
4. Accertarsi che la pompa sia posizionata in modo stabile. Verificare che la pompa sia fissata tirandola via con delicatezza dalla stazione
di aggancio/workstation* senza utilizzare la leva di rilascio. Se la pompa è collegata in modo sicuro, non si deve staccare dalla
stazione di aggancio/workstation*.
5. Per rimuovere la pompa, spingere la leva di sgancio e tirare in avanti la pompa.
Se non montata correttamente, la pompa potrebbe cadere dalla stazione di aggancio/workstation* con il rischio di
lesioni per l'utente e/o il paziente.
Barra
rettangolare
Camma girevole
Leva di sgancio (premere per rilasciarla)
*Alaris DS Docking Station e Alaris Gateway Workstation.

BDDF00638 Edizione 2
18/56
Pompa a siringa Alaris™ PK Plus
Caricamento della siringa
Caricamento della siringa
Preparazione di siringa e set di somministrazione
Per ridurre potenziali ritardi all'avvio, imprecisioni di somministrazione e generazione ritardata degli allarmi di occlusione, a ogni
caricamento di una nuova siringa:
• Utilizzare la siringa di dimensioni più piccole possibili; ad esempio, per l'infusione di 9ml di fluido utilizzare una siringa da 10ml.
• Utilizzare l'opzione SPURGO SIRINGA o SPURGO sulla Pompa per ridurre il ritardo nell'avviamento dell'infusione; consultare la
sezione Avvio della pompa.
Utilizzare la siringa dalle dimensioni minime compatibili necessarie per erogare il fluido o il farmaco; tale aspetto è
particolarmente importante per l'infusione di farmaci ad alto rischio o vitali a basse portate, soprattutto inferiori a
0,5ml/h.
Spurgare il sistema della Pompa prima di avviare un'infusione o dopo la sostituzione di una siringa quasi vuota con
una siringa di ricambio. Durante lo spurgo, verificare che il set di prolunga non sia collegato al paziente.
Raccomandazioni di procedura clinica:
• Diametro interno del tubo: per infusioni a basse velocità si consiglia l'uso di un tubo microbore o con diametro ridotto
• Filtri: volume interno e spazio morto dei filtri in linea devono essere ridotti al minimo
• Siti di connessione: i farmaci critici devono essere collegati il più vicino possibile al sito di accesso vascolare
Posizionamento della pompa
Verificare che la Pompa si trovi il più vicino
possibile al livello del cuore del paziente.
Il livello del cuore del paziente dovrebbe
essere in linea con la parte centrale della
Pompa.
La regolazione dell'altezza della Pompa rispetto al livello del cuore del paziente può portare a un aumento o
diminuzione temporanei nell'erogazione del fluido.
w
Se si utilizzano pompe a siringa multiple e se non è possibile dal punto di vista clinico posizionarle tutte al livello del
cuore del paziente, posizionare i farmaci vitali o ad alto rischio il più vicino possibile al livello del cuore del paziente.
w
Durante l'infusione di farmaci vitali o ad alto rischio, pensare di posizionare le Pompe in infusione alle velocità più
basse il più vicino possibile al livello del cuore del paziente.

BDDF00638 Edizione 2
19/56
Pompa a siringa Alaris™ PK Plus
Caricamento della siringa
Caricamento e conferma di una siringa
Per caricare in modo stabile una siringa e confermare l'operazione, seguire i passaggi indicati di seguito. Un
caricamento incorretto della siringa può comportare un'errata identificazione del tipo e delle dimensioni della
siringa. Una tale evenienza potrebbe comportare una velocità di infusione imprecisa e compromettere le prestazioni
della pompa.
Usare solo siringhe del tipo indicato sulla pompa o nel manuale. L’uso di una siringa non presente nell'elenco può
influire sulla precisione della velocità di infusione e compromettere il funzionamento della pompa.
Nel caricare inizialmente il fluido nella siringa, occorre tenere conto del volume del liquido che rimane all'interno
dello "spazio morto" del set prolunga e della siringa al termine dell'infusione, poiché questo fluido non viene infuso.
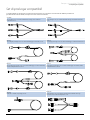
Morsetto di fissaggio della flangia della siringa
Morsetto
blocca siringa
Flangia del
cilindro
Fermi dello
stantuffo
Flangia dello
stantuffo
Porta-
stantuffo
Plunger
Impugnature
sagomate
Cilindro
della siringa
Posizionare la Pompa su una superficie orizzontale stabile o fissarla come descritto in precedenza.
Collocare la pompa su una superficie orizzontale stabile o fissarla come descritto in precedenza. Preparare, caricare ed eseguire il
riempimento della siringa e della prolunga monouso utilizzando le normali tecniche di asepsi.
1. Premere l'impugnatura sagomata porta-stantuffo e far scivolare il meccanismo a destra.
2. Tirare il morsetto blocca siringa in avanti, quindi spingerlo verso il basso.
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
La pagina si sta caricando...
-
 1
1
-
 2
2
-
 3
3
-
 4
4
-
 5
5
-
 6
6
-
 7
7
-
 8
8
-
 9
9
-
 10
10
-
 11
11
-
 12
12
-
 13
13
-
 14
14
-
 15
15
-
 16
16
-
 17
17
-
 18
18
-
 19
19
-
 20
20
-
 21
21
-
 22
22
-
 23
23
-
 24
24
-
 25
25
-
 26
26
-
 27
27
-
 28
28
-
 29
29
-
 30
30
-
 31
31
-
 32
32
-
 33
33
-
 34
34
-
 35
35
-
 36
36
-
 37
37
-
 38
38
-
 39
39
-
 40
40
-
 41
41
-
 42
42
-
 43
43
-
 44
44
-
 45
45
-
 46
46
-
 47
47
-
 48
48
-
 49
49
-
 50
50
-
 51
51
-
 52
52
-
 53
53
-
 54
54
-
 55
55
-
 56
56
-
 57
57
-
 58
58


























































BD Pompa a siringa Alaris™ PK Plus MK4 Istruzioni per l'uso
- Tipo
- Istruzioni per l'uso
Documenti correlati
-
BD Pompa a siringa Alaris™ PK Plus MK4 Istruzioni per l'uso
-
-
-
-
-
-
-
-
-